



Does the Amyloid Model of Alzheimer’s Disease Still Make Sense? My Journey to Resolve the Gaps
Whoever can pinpoint the missing piece will be on the verge of solving this seemingly unsolvable disease.

This article was originally published at Microbial Instincts on Oct 14, 2024.

When I was a final-year undergraduate student learning about advanced neurobiology in 2019, I was taught that Alzheimer’s disease (AD) is caused by excessive accumulation of amyloid-beta (Aβ) until it forms toxic plaques in the brain.
I accepted that as a textbook fact. After all, it makes sense that toxic plaques in the brain would trigger neurodegeneration, and, of course, clearing them would solve the problem.
But five years later, after immersing myself in research rather than textbooks, I learned that the amyloid hypothesis of AD isn’t the full story. I learned that science can change and evolve, and sometimes, it takes a 180° flip in reasoning to answer seemingly unsolvable questions.
Other scientists have raised similar doubts about the validity of the amyloid hypothesis of AD in academic papers (Figure 1). In this article, I will re-convey their message, as well as others, in a more comprehensible blog post while also adding my own insights into the matter.
Here’s my journey to pinpoint and resolve the gaps in the current model of AD. It’s a 30-minute read that took me months (part-time) to research, think, and write. I hope you will find it as worthwhile as I do.
I’ll first describe (i) the huge disappointment of current anti-Aβ drugs to (ii) why the amyloid hypothesis of AD was so convincing and the problems with this hypothesis, and, finally, to (iii) the alternative model of AD.

1. The huge disappointment of anti-Aβ drugs
1.1. Current landscape
At present, cholinesterase inhibitors are the standard practice in managing AD symptoms. These include donepezil, rivastigmine, and galantamine, which were approved by the FDA between 1996 and 2001.
They increase levels of acetylcholine, a neurotransmitter vital for learning and memory, in the brain. As they don’t target the underlying cause of AD — believed to be Aβ aggregation — these drugs are not cures. They only work temporarily and are not viable long-term solutions.
As a result, tremendous efforts have been dedicated to designing anti-Aβ drugs. After decades, the Food and Drug Administration (FDA) has recently approved three anti-Aβ drugs for AD:
Aducanumab (tradename: Aduhelm) by Biogen and Eisai.
Lecanemab (tradename: Leqembi) by Biogen and Eisai.
Donanemab (tradename: Kisunla) by Eli Lilly and Company.
All are anti-Aβ antibodies designed to bind to and clear Aβ from the brain, although they target different stages of Aβ formation (Figure 2). But many experts have questioned the clinical effectiveness and safety of these three drugs, which, combined with their unaffordable costs, further tarnishes the reputation of the amyloid hypothesis of AD.

1.2. Aducanumab
Let’s start the story with aducanumab.
In 2021, the Food and Drug Administration (FDA) approved aducanumab via a fast-tracked approval process for drugs that fill an unmet medical need. That time was met with hype and optimism as aducanumab was the first approved Aβ-targeting drug in history, ever since the amyloid hypothesis of AD became the consensus model in the 1990s.
Since then, between 2004 and 2021, at least 550 phase II/III clinical trials on AD have been conducted, examining at least 98 compounds, most of which targeted Aβ in the brain. Only two compounds managed to pass phase III clinical trials, giving a 2% success rate. Of the two, only one — aducanumab — successfully gained FDA approval. (The other compound is an algae extract, which was approved in China and not other countries due to unconvincing and potentially fraudulent clinical data.)
So, it took about two to three decades of intense research into the amyloid hypothesis of AD to develop a successful anti-Aβ drug, a success that would later become the greatest blunder of the decade, however.
Apparently, the FDA approved this drug based on the surrogate endpoint of reduced Aβ content in the brain. In clinical trials, a surrogate endpoint is a substitute indicator of the drug’s effect on a true clinical outcome, which should be cognitive improvement in AD. But the caveat is that Aβ reduction doesn’t necessarily improve symptoms.
In fact, Biogen and Eisai previously announced they were stopping the trials after preliminary analyses indicated aducanumab did not improve symptoms of AD patients. But 6 months later, Biogen (Eisai had given up on aducanumab) reversed their decision by presenting a subgroup analysis showing the drug slightly reduced cognitive decline and brain Aβ levels in patients who received higher doses of the drug.
But doing subgroup analysis when the main analysis is null is not advisable because it risks false positives. The smaller sample size in subgroups can lead to less reliable results, so any benefit in subgroups should be interpreted cautiously rather than seen as definitive evidence of benefit.
A cautionary incident is the 2016 clinical trial involving solanezumab, another anti-Aβ drug. While the main analysis found no benefit, subgroup analyses suggested that the drug lowered cognitive decline by 34% in those with mild AD. This prompted a more extensive trial in 2018, which included only participants with mild AD — but it ultimately failed to demonstrate a significant benefit, disappointing many.
Moreover, an independent advisory committee voted 8:1 that the data from the subgroup analysis did not show “primary evidence of effectiveness.” The FDA doesn't usually go against the independent advisory panel, but they did it for aducanumab based on “tremendous unmet medical need” for AD treatments. One FDA adviser described the drug’s approval as “the worst approval decision that the FDA has made that I can remember.” Three FDA advisers even resigned after the drug’s approval.
“The FDA’s decision to approve aducanumab for anyone with Alzheimer’s disease, regardless of severity, showed a stunning disregard for science, eviscerated the agency’s standards for approving new drugs, and ranks as one of the most irresponsible and egregious decisions in the history of the agency.”
— Michael A. Carome, MD
The high price tag of US$56,000 per year did not help the drug’s case. Medicare insurance refused to cover aducanumab for patients outside of the trial. Physicians were also wary of prescribing this drug due to its lack of efficacy and safety concerns. As a result, aducanumab had terrible sales, leading Biogen to abandon the drug in January 2024.
1.3. Lecanemab
The story doesn’t end with aducanumab, however.
Biogen and Eisai also have another anti-Aβ antibody, lecanemab, which also has its fair share of drama and controversy.
Like aducanumab, lecanemab received FDA approval via a fast-tracked process in 2023, marking the second anti-Aβ drug to be approved. But like aducanumab, lecanemab’s efficacy data is unconvincing.
At first glance, lecanemab’s phase III clinical trial showed a promising 27% decline in cognitive decline among patients with early-stage AD. At a second glance, however, the 27% decline is relative; in absolute terms, lecanemab only slowed cognitive decline by 0.45 points on the 18-point CDR-SB (clinical dementia rating — sum of boxes) scale.
This benefit is below the minimal clinically important difference, which should be at least 1 point on CDR-SB for mild AD. So, the statistically significant efficacy of lecanemab is not clinically meaningful.
To understand the difference between relative and absolute increase, consider an intervention that reduces the incidence of x from 2% to 1%. In this case, the relative reduction would be (1%/2% = 50%), while the absolute reduction is a mere 1% (2%–1% = 1%).
1.4. Donanemab
Moving on to donanemab, this one is probably the least bad.
Donanemab was just approved by the FDA in July 2024 based on its phase III trials, which showed an 80–85% reduction in brain Aβ content, a 35% reduction in cognitive symptoms, and a 39% reduced risk of disease progression in AD patients with early signs of Aβ aggregation.
That sounds amazing, except it suffers the same issue as lecanemab regarding statistical significance vs. effect size. Specifically, donanemab only conferred a 0.7-point reduction in the CDR-SB scale.
Remember, at least 1 point in the CDR-SB scale is needed for minimal clinical efficacy. So, while donanemab fared better than lecanemab (0.7- vs. 0.35-point reduction), it’s still inadequate to meet the minimal clinical efficacy of at least a 1-point change in the 18-point CDR-SB scale.
But the independent advisory committee voted positively for donanemab — a good surprise since the FDA went against the committee for aducanumab and did not consult the committee for lecanemab. That said, the committee also cautioned that more research is needed to understand who would benefit from donanemab and how long the benefit would last, as well as balancing the drug’s potential benefits with its potential risks.
“The efficacy of these drugs is not translated into improvements,” Alberto Espay, MD, a professor of neurology at the University of Cincinnati, Ohio, said about the recent anti-Aβ drugs. “They just mean a statistically significant but clinically meaningless slower decline.”
1.5. Amyloid-related imaging abnormalities (ARIA)
Speaking of risks, these anti-Aβ drugs are not entirely safe.
They come with a rare risk of amyloid-related imaging abnormalities (ARIA), which has been seen across anti-Aβ drug trials:
In aducanumab’s trial, ARIA occurred in 35–40% of the high-dose group and only 1–10% in the placebo group; about 1% of ARIA victims in the high-dose group suffered severe neurological symptoms.
In lecanemab’s trial, ARIA occurred in 21% of the patients taking the drug compared to only 9% of those on a placebo. While most cases were harmless, some patients became seriously ill, and at least two patients died from brain swelling and bleeding.
In donanemab’s trial, symptomatic ARIA occurred in 24% of the participants in the drug group but only 2% in the placebo group. Among these cases, 9% and 0.5% in the drug and placebo group had severe neurological symptoms that led to trial discontinuation.
The FDA was aware of this safety signal but approved those anti-Aβ drugs based on the justification that they met the goal for efficacy — an efficacy that is minuscule at best and negligible at worst.
“You’re dealing with people with mild cognitive impairment who are functioning, and you’re putting them at risk,” Diana Zuckerman, PhD, president of the U.S. National Center for Health Research, said in response to lecanemab’s approval, and the same thing can be said for the questionable approvals of aducanumab and donanemab.
Overall, it’s clear that these anti-Aβ drugs lack convincing clinical effectiveness and carry a small risk of severe neurological harm. But why? If the amyloid hypothesis of AD holds true, why are no anti-Aβ drugs tested over the past two decades ever truly successful?
Several reasons have been put forth. These include difficulties in recruiting the right participants who would benefit from the drug and designing the right trials to show that the drug can work in specific circumstances. Notwithstanding these methodological reasons, another, perhaps more obvious, reason is that the amyloid hypothesis of AD is incomplete.
This [referring to repeated failed anti-Aβ drugs in clinical trials] gives AD research a bad name. This is not evidence-based science. This is amyloid hypothesis driven theology. We can only imagine the people living with AD having volunteered for the clinical trials, their family members, friends. and other carers, their hopes and dreams all but dashed. How long do the people living with AD have to wait? There are no AD survivors.
— Markku Kurkinen, PhD
2. Why the amyloid hypothesis was so convincing
2.1. The origin
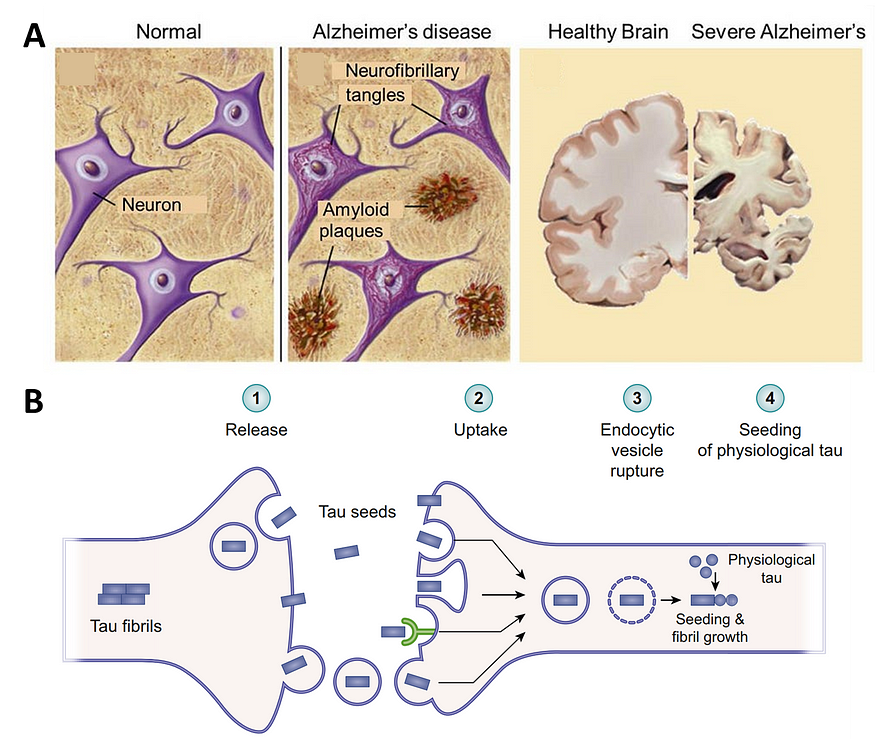
In 1906, Alois Alzheimer, a German psychiatrist and neuroanatomist, discovered the presence of amyloid plaques and neurofibrillary tangles (NFTs) during the brain autopsy of a patient suffering from severe memory loss, a disease that would later be called Alzheimer’s disease (AD).
(NFTs are composed of aggregated tau proteins, which represent the second hallmark pathology of AD after Aβ plaques.)
But it wasn’t until 1987 that scientists identified and sequenced the gene of amyloid precursor protein (APP) — whose incorrect processing could accumulate Aβ plaques — in chromosome 21 of the human genome. This finding was pivotal because individuals with Down syndrome, who have an extra copy of chromosome 21, often develop AD at an early age — suggesting a direct link between APP, Aβ, and AD.
This link became even stronger in 1991 when scientists discovered that mutations in the APP gene caused early-onset familial AD, an inherited form of AD that occurs at a young age of the 30s or 40s rather than typical non-inherited AD at the age of 60 and above. A mutated APP gene was found to increase the production of altered amyloid, particularly amyloid-beta 42 (Aβ42) that’s prone to aggregation and plaque formation.
Around the same time, mutations in the presenilin genes, which are involved in APP processing, were also linked to early-onset familial AD. These genetic findings indicated that Aβ accumulation was not just a byproduct of the disease but a driving force in its development.
Based on these discoveries, the “amyloid cascade hypothesis” was proposed in a 1991 landmark paper — stating that Aβ accumulation in the brain is the initial trigger for a cascade of events leading to AD, including NFTs, neuronal death, and cognitive decline (Figure 3).
Further research showed that brain Aβ content correlated with the risk of developing AD and the rate of disease progression in humans. Causative evidence can be derived from animals, where the artificial introduction of Aβ plaques triggered AD-like behavior.

The amyloid hypothesis quickly became the dominant model in AD research, guiding drug development for decades. Many treatments were designed to reduce Aβ production or clear existing plaques based on the belief that doing so could halt or slow AD progression.
But despite extensive research and numerous clinical trials, no Aβ-targeting drugs have been truly successful in showing meaningful clinical benefits, leading to growing debate about the amyloid hypothesis.
Critics argue that while Aβ does play a role in AD, it may not be the sole cause, and it may not even be the enemy we thought it was. Before we delve into other causes of AD and re-consider the role of Aβ, let’s understand where the amyloid hypothesis fell short.
[A] legitimate concern is that the hypothesis has become ‘too big to fail’. With so much time, money and, indeed, faith invested in the construct, is a negative outcome simply intolerable for the scientific community and society who depends on it? Moreover, does expansion of trials run the risk of obtaining a kernel of positive data, purely out of randomness and the expanding denominator, thus perpetuating a fundamentally flawed paradigm and diverting attention from biological processes more worthy of targeting?
— Rudy J. Castellani, MD, and Mark A. Smith, PhD.
2.2. Problems with the amyloid hypothesis
Evidence opposing the amyloid hypothesis surfaced when autopsy investigations found that about 10–50% of people without AD also exhibit signs of moderate-to-high Aβ aggregation that would qualify them for an AD diagnosis.
Scientists tried to explain this discrepancy with the concept of cognitive reserve, which refers to the brain’s ability to use alternative networks or processes to compensate for damage and maintain function. Factors such as higher education, mental stimulation, and social engagement can strengthen cognitive reserve, allowing some people to resist the damaging effects and clinical symptoms of Aβ plaques. Still, cognitive reserve is not a definitive explanation and remains a hypothesis to account for the variation in how individuals experience cognitive decline.
That said, more evidence against the amyloid hypothesis dates back to, ironically, the year the hypothesis was proposed in 1991.
A 1991 study mapped the distribution of Aβ plaques and NFTs across 39 brain areas of AD patients. Oddly, they found that plaques “were more evenly distributed throughout the cortex, with the exceptions of limbic periallocortex and allocortex, which had notably fewer [plaques] than other cortical areas.” This is weird because the allocortex is the brain’s memory region, where the hippocampus is located.
Instead, the 1991 study found that the “limbic periallocortex and allocortex had more NFTs than any other type of cortex.” Subsequent efforts also confirmed the presence of NFTs in the hippocampal regions of cases of early-stage AD (Figure 4), leading to the tau hypothesis of AD.

2.3. Problems with the tau hypothesis
Tau proteins can get phosphorylated excessively so that they start to self-aggregate into neurotoxic NFTs. As evidence of early NFT formation was found in the brain memory regions before Aβ plaques, it lends credence to the hypothesis that tau is the pathogenic trigger of AD rather than Aβ.
Scientists generally agree that NFTs better correlate with the onset and severity of AD than Aβ plaques. The brain distribution pattern of NFTs also tracks better with the progressive stages of AD (Figure 4). Further, in the final stages of AD, the brain memory regions were most severely affected by the increased NFT formation rather than Aβ plaques.
More evidence favoring the tau hypothesis is its higher neurotoxicity. Unlike Aβ plaques that form outside neurons, NFTs form inside the neurons (Figure 5A), directly triggering neurodegeneration. Misfolded tau can also be released and taken up by nearby neurons due to natural cellular processes like nutrient uptake and debris clearance. Once inside, these tau seeds further trigger NFT formation. This process allows tau aggregation to propagate across neurons and brain regions (Figure 5B).
However, the tau hypothesis of AD is not without problems.
First, there’s a lack of causative genetic evidence for tau compared to the strong genetic case of Aβ in causing familial (inherited) AD. While mutations in the tau-encoding gene can cause frontotemporal dementia with Parkinsonism, this is not the same disease as AD. This fact is also a huge blow to the tau hypothesis of AD as it implies that tau could be the key pathogenic trigger of a different form of dementia that’s not AD.
Second, tau neurotoxicity seems to be dependent on Aβ aggregation. Mutating the APP (amyloid precursor protein) gene in addition to the tau gene was necessary to induce the formation of NFTs in the brain memory regions. Moreover, efforts to remove Aβ plaques have resulted in reduced NFTs in mice. This causative evidence in animals suggests that tau is secondary to Aβ and that Aβ is vital for the tau hypothesis to work.
(While animal models are not entirely representative of humans, fundamental pathological processes should be reproducible in simpler living models like mice. Put another way, if the hypothesis doesn’t work in simpler models, it’s very unlikely to work in humans.)
Tau and Aβ toxicity may also depend on each other. Studies showed that Aβ activates enzymes that hyperphosphorylate tau into NFTs, which then gain a greater affinity for Fyn. This protein is important for NMDA receptors to mediate Aβ plaque’s neurotoxicity. As a result, tau and Aβ both fuel each other’s aggregation, creating a vicious cycle that drives AD.
Third, as with Aβ-targeting drugs, tau-targeting drugs have not been successful in clinical trials either. For example, one target of interest is glycogen synthase kinase 3 beta (GSK-3β), a protein kinase that promotes tau phosphorylation. But Tideglusib, a GSK-3β inhibitor, failed to show clinical benefits as early as in the phase II clinical trial. Likewise, several anti-tau antibody therapies have failed phase II clinical trials.
While methylene blue derivatives were claimed to show potential benefits in slowing cognitive decline in phase III trials by inhibiting tau, the trial methods and therapy effectiveness remain controversial. Much like anti-Aβ drug trials (as discussed above), they rely on subgroup analyses with questionable effect sizes due to negative primary analyses.
2.4. The emergence of other hypotheses
As both the amyloid and tau hypotheses of AD appear incomplete, scientists have theorized several alternative hypotheses over the years. At least 10 of them have been proposed:
Mitochondrial Cascade: Mitochondrial efficiency, influenced by genetics and environmental factors, determines how aging affects cells. As mitochondria become less efficient with age, they contribute to cellular damage, APP misprocessing, and Aβ plaque accumulation, leading to or accelerating AD pathology.
Oxidative Stress: Mitochondria are the primary producers of reactive oxygen species (ROS), and in AD, ROS production increases due to mitochondrial damage. The imbalance between ROS and antioxidants leads to oxidative stress, which could damage neurons.
Epigenetic Dysregulation: Mitochondrial metabolites, like α-ketoglutarate and oxaloacetate, are essential for epigenetic modifications. Disruptions in mitochondrial function alter the production of these metabolites, leading to faulty epigenetic regulation and abnormal gene expression in neurons.
Mitophagy Impairment: Mitophagy is the process by which cells remove damaged mitochondria. In AD, mitophagy is impaired, leading to the accumulation of dysfunctional mitochondria and exacerbating neurodegeneration.
Calcium Homeostasis: Aβ plaques can disrupt calcium balance within neurons. This makes them more prone to damage from environmental stimuli and contributes to excitotoxicity, neuronal death, and loss of synaptic plasticity, which is crucial for learning and memory.
Neurovascular Impairment: Vascular dysfunction in the brain impairs blood flow, reducing oxygen and nutrient delivery to neurons. Aβ plaque can also damage cerebral arteries and the blood-brain barrier (BBB), leading to neurovascular uncoupling. This process can aggravate brain hypoxia (low oxygen) and inflammation.
Inflammation: Aβ plaques trigger sustained inflammatory responses, releasing cytokine that damages neurons. Inflammation further exacerbates the buildup of Aβ plaques and NFTs, creating a vicious cycle of neuro-inflammation and -degeneration.
Insulin Dysregulation: AD shares similarities with type 2 diabetes, particularly through cerebral insulin resistance. In AD, neurons become inflamed and resistant to insulin, disrupting normal cell functions.
Metal Ion Dysregulation: Biometals, particularly copper, zinc, and iron, play a key role in Aβ aggregation and neurotoxicity. These metals can bind to Aβ, promoting plaque formation and oxidative stress. Excess iron, in particular, generates toxic free radicals that contribute to neuronal damage, further driving AD pathology.
Lymphatic System Impairment: The brain’s lymphatic system is responsible for clearing waste products, including Aβ plaques, from the brain. The impaired function of this system leads to the accumulation of toxic substances in the brain.
But one fundamental flaw is that while these hypotheses play a role, they are not unique to AD. Basic cellular dysfunction involving mitochondria, oxidative stress, inflammation, insulin, and metal ions occur in numerous other diseases. In other words, these are basic signs of a disease.
2.5. The missing piece
To truly understand the cause of AD, we must first identify the initial trigger behind its hallmark features — Aβ plaques and NFTs. An effective model of AD needs to explain the development of these two fundamental pathological markers, starting from the hippocampus.
Put simply, while the amyloid and tau hypotheses of AD are not complete, we cannot deny that Aβ plaques and NFTs are unique signs† of AD. But treating these unique signs doesn't address the underlying cause or trigger, which may explain why AD drug trials have failed miserably.
†They are not exactly unique, as they can also appear in a few other neurodegenerative diseases (i.e., Aβ plaques in cerebral amyloid angiopathy and Down’s syndrome; NFTs in Pick’s Disease and progressive supranuclear palsy), but they are close enough.
So, what exactly triggers the formation of plaques and tangles, starting from the hippocampus? While the precise trigger remains uncertain, we can make inferences based on what we already know.
First, Aβ plaques are formed due to the abnormal processing of amyloid precursor protein (APP), and NFTs result from the abnormal phosphorylation of tau proteins. Next, we examine the factors that could disrupt APP processing and tau phosphorylation. These include inflammation, oxidative stress, and mitochondrial dysfunction — all of which are common signs of cellular dysfunction.
But this is where things get complicated. If inflammation or oxidative stress alone caused AD, anti-inflammatory and anti-oxidative treatments would have succeeded in curing the disease by now. This suggests that there is an additional factor that triggers both the pathological hallmarks of AD and these basic signs of disease.
There must be something else that triggers the cascade of cellular dysfunction that leads to AD specifically — with its plaques and tangles — and not other diseases like heart diseases or cancer.
Identifying this missing piece is what we need in the field of AD.
Many researchers, including myself, suspect that one of the missing pieces is an infectious agent, specifically herpesviruses.
A disclaimer is that I’ve written extensively on the infectious origin of neurodegenerative diseases and published academically on it. So, I admit I could be biased, but let me outline the evidence and discuss the caveats as objectively as I can.
3. An alternative theoretical model for AD
3.1. Reverse-causation
Let’s start with a reverse cause-and-effect argument.
Fascinating data comes from a natural experiment in the U.K. involving the live herpes zoster (shingles) vaccine. This 2023 study leveraged a unique situation where individuals born before September 2, 1933, were ineligible for the live shingles vaccine, while those born on or after that date were eligible. This setup provided a naturally randomized cohort that allowed researchers to compare dementia rates between the two groups, free from many biases that often confound observational studies.
(The act of randomization evenly distributes the countless number of variables that are different between individuals, ensuring that the outcome seen is strictly due to the intervention, i.e., live shingles vaccination, thereby establishing cause and effect.)
“[Apart from] the probability of ever receiving the herpes zoster vaccine, there is no plausible reason why those born just one week prior to 2 September 1933 should differ systematically from those born one week later,” the study authors said. “This unique natural randomization, thus, allows for robust causal, rather than correlational, effect estimation.”
Over seven years, the vaccine-eligible group had a 20% reduced incidence of dementia specifically compared to the ineligible group. The incidence of other diseases (e.g., cancer and diabetes) did not change, indicating that the protective effects of shingle vaccination are specific to dementia. The study also ruled out confounding factors (e.g., healthcare access or other vaccinations), demonstrating a specific causal link between the live shingles vaccine and reduced dementia risk.
(Note: the most common form of dementia is AD.)
Importantly, similar results were recently replicated in two studies from Australia and the U.S. in 2024.
The Australian study also capitalized on the eligibility cutoff date for receiving the live shingles vaccine, revealing that those eligible had a 2% point reduction in dementia diagnoses (from about 6% to 4%) over 7.4 years (Figure 6A). Crucially, eligibility for the vaccine did not affect the likelihood of receiving other healthcare services, including other vaccines, nor did it influence the incidence of other chronic conditions beyond dementia (Figure 6B) — strengthening the fact that shingles vaccination specifically protects against dementia in a causal manner.
Using a similar natural randomization setting, the U.S. study reported that another type of shingles vaccine — i.e., protein recombinant — was 17% more effective than the live one in lowering the risk of dementia. But both vaccines were 15–25% more effective at reducing dementia risk than influenza or tetanus-diphtheria–pertussis (Tdap) vaccines.
These findings are profound.
If two types of shingles vaccines can prevent or delay dementia in a cause-and-effect manner, as demonstrated by three studies from different countries, it provides compelling reverse-causation evidence that herpes zoster could trigger the development of AD, at least in certain cases.

3.2. The most suspicious herpesvirus
Now, what’s herpes zoster actually? Herpes zoster virus, also known as human herpesvirus 3, first causes chickenpox. Then, it stays dormant in neurons and may reactivate in later life to cause shingles (painful rash) under conditions of immunosuppression or stress.
Among the 8 types of herpesviruses known to infect humans, we only have an approved vaccine for herpes zoster. But it’s actually herpes simplex virus 1 (HSV-1) — a virus that can cause mild oral sores or encephalitis — that has been widely linked to AD in the academic literature.
Specifically, HSV-1 has been found to:
Reproduce plaques and tangles, causing AD/dementia in animals, as well as lab-cultured neurons and human mini-brains.
Be present and co-localize with Aβ plaques in brain autopsies of patients who died from AD/dementia.
Increase the risk of AD/dementia in humans in observational studies, especially for individuals displaying signs of HSV-1 reactivation, such as active IgM antibodies and the appearance of oral sores.
Although the evidence mainly identified HSV-1’s contribution to dementia, herpes zoster virus also plays a role.
A 2022 study found that herpes zoster infection alone did not trigger Aβ aggregation in neurons, while HSV-1 infection did. But in the presence of HSV-1 dormancy, herpes zoster infection triggered HSV-1 reactivation and more Aβ aggregation than HSV-1 infection alone.
“It’s a one-two punch of two viruses that are very common and usually harmless, but the lab studies suggest that if a new exposure to [VZV] wakes up dormant herpes simplex virus, they could cause trouble,” said Dana Cairns, Ph.D., a biomedical engineer, and the study’s lead author.
If herpes zoster vaccination is enough to prevent AD/dementia in a causal manner, imagine what an HSV-1 vaccine would do.
Unfortunately, HSV-1 is one of the very tricky viruses to immunize against, such as human immunodeficiency virus and hepatitis C virus. As a result, all the prior vaccine trials for HSV-1 have failed.
That said, antiviral drugs targeting HSV-1 have shown promise in protecting against AD/dementia in observational studies. For example, in a 2018 nationwide study in Taiwan, those who had HSV-1 infection had a 2.5-fold increased risk of dementia. But anti-herpes drugs lowered this risk by 90% compared to no drugs. In a more recent large cohort study in Sweden, those who had HSV-1 or other herpes infections but did not take antivirals had a 1.5-fold increased risk of dementia; this risk was not only nullified in those who took anti-herpes drugs but also fell by 10%.
As such, an ongoing clinical trial is testing the efficacy of valacyclovir, an anti-herpes drug, in treating mild AD patients. Results will likely come out in 2025. At least for now, a 2024 pilot clinical trial showed that short-term treatment with high-dose valacyclovir was safe and tolerable in a small group of early AD patients with HSV-1 seropositivity, who also showed a slight improvement in cognitive scores after treatment.
But scientists have expressed reservations. For instance, it’s unclear if antivirals would provide much clinical benefit once symptoms have begun, as this means the trigger — i.e., HSV-1 triggering plaques and tangles — has been pulled. Additionally, antivirals can’t eradicate HSV-1 since HSV-1 can re-enter a dormant state, with the capacity to reactivate later, questioning if lifelong antivirals would be necessary to curb AD.
3.3. Decoding the mechanisms
Unbeknownst to many, a wealth of research has been done to decode the mechanisms of how HSV-1 can cause — or, at least, contribute to — AD.
I’ve published academically about this in my 2021 paper, “The Hippocampal Vulnerability to Herpes Simplex Virus Type I Infection: Relevance to Alzheimer’s Disease and Memory Impairment.” As the title implies, the uncanny role of HSV-1 in AD development lies in the virus’s tropism† to infect the hippocampus, the memory region in the brain that first begins to fail in AD due to the development of plaques and especially tangles.
(†Medically, tropism refers to the capacity of a pathogen to infect and target a particular area or tissue within the body.)
Researchers have done extensive experiments to map out the infection pathways of HSV-1 (Figure 7). HSV-1 can infect the human body via three routes: the mouth, nose, and eyes. Regardless of the entry route, HSV-1 first seeks the trigeminal ganglion (a bunch of nerves near the base of the brain) to establish dormancy. The olfactory bulb in the brain could also be the site of HSV-1 dormancy, but this is optional.
From the trigeminal ganglion (and/or olfactory bulb), HSV-1 can reactivate under conditions of stress or immunosuppression to infiltrate the brain. Specifically, HSV-1 travels up the brainstem and always infects the hippocampus first. The further spread of HSV-1 to other brain regions can cause brain inflammation, i.e., herpes simplex encephalitis (HSE).
In fact, it was the observation that HSE affects similar brain regions as AD that led Melvyn J. Ball, MD, a neuropathologist, to hypothesize that HSV-1 reactivation could induce sub-clinical neuronal damage that, when accumulated over time, leads to AD in as far as back in 1982.

But what makes the hippocampus particularly vulnerable to HSV-1 infection and not the other brain regions?
In my 2021 paper, I proposed 5 key arguments for this:
High expression of cellular receptors for HSV-1: HSV-1 entry into cells begins when its glycoproteins bind to specific receptors on the cell surface. Compared to other brain regions, transcriptomic analyses found that the hippocampus has 2–3 times higher expression of molecules (e.g., nectin-1, HVEM) that HSV-1 can use as receptors.
Abundance of neurogenic stem cells: As a brain region responsible for memory formation, the hippocampus is rich in stem cells. HSV-1 and other viruses tend to infect these stem cells due to their higher rate of cell division and higher expression of heparan sulfate proteoglycans (HSPGs), another receptor that HSV-1 can use to infect cells.
Low anti-viral immunity: The hippocampus has lower levels of anti-viral immune factors such as interleukin-6 (IL-6) and relies on microglia (immune cells of the brain) for immune defense. However, the hippocampus is especially susceptible to age-related decline in microglial function and anti-viral immunity.
High expression of glucocorticoid receptors (GRs): These receptors bind to stress hormones like cortisol. The hippocampus has high levels of GRs compared to other brain regions, making it particularly sensitive to stress-related pathology and stress-induced reactivation of HSV-1.
High expression of amyloid precursor protein (APP): The hippocampus expresses high levels of APP, which HSV-1 can exploit for viral transport. HSV-1 infection has been shown to trigger abnormal APP processing, leading to increased Aβ accumulation.
All these factors make the hippocampus the prime target of HSV-1 infection. As the hippocampus mediates memory formation, it’s one of the earliest regions to be affected by AD. It’s also one of the most severely affected brain regions by the neurodegenerative effects of AD.
But I’m not the first to put forward the hippocampal vulnerability to HSV-1 as a potential trigger of AD. As mentioned, Dr. Ball is the pioneer of this hypothesis, where HSV-1 might induce neuron-to-neuron tauopathy and Aβ spread in AD as HSV-1 propagates along its infection pathways, starting from the hippocampus (Figure 8). Dr. Ball also emphasized that pinpointing the biological basis behind the hippocampal origin of AD could be the key to solving this neurodegenerative disease.
Once we can explain why neurofibrillary tangles always make their [brain] debut specifically here, in the hippocampal formation, we will be within reach of unraveling the etiology of AD. — Melvyn J. Ball (2006).
Whatever the eventual explanation for the focality of such [hippocampal] lesions in the mesial temporal lobe, those researchers who discover the biological basis for this predilection and focal selectivity will then be within grasp of comprehending the essential etiology for this devastating neurodegenerative disorder.— Melvyn J. Ball (1988).

Indeed, modern research has shown that HSV-1 infection in the hippocampus alone could trigger the formation of plaques and tangles, as well as neuroinflammation and oxidative stress, in animals and lab-cultured human mini-brains — essentially reproducing the pathological hallmarks of AD. Even though these are conducted in laboratory settings, they are the best we can do. After all, it’s unethical to deliberately infect people with HSV-1 to see if it causes plaques and tangles.
The precise mechanisms by which HSV-1 could trigger plaques and tangles are complex. But, in general, they involve the pathological interactions between HSV-1 glycoproteins and cellular processes that mediate APP processing and tau phosphorylation, as well as oxidative stress, autophagic dysfunction, and neuroinflammation (Figure 9).
Coupled with epidemiological evidence showing that HSV-1 reactivation — indicated by the appearance of oral sores or IgM antibodies — does increase the risk of AD/dementia, it becomes difficult to deny the involvement of HSV-1 in triggering this neurodegenerative disease.

3.4. Pros of the infectious model of AD
The infectious model of AD actually answers several gaps in the current amyloid paradigm of AD. First, it could explain the hippocampal origin of AD, or else the classic amyloid model does not specify the precise trigger of the first plaque or first tangle in the hippocampus.
Second, the infectious model of AD complements the genetic model of AD. We know that the strongest genetic risk factor for AD is the APOE4 gene. Compared to other APOE genes, APOE4 is less efficient at lipid metabolism and Aβ clearance. Those with one copy of this gene have a 4-fold increased risk of AD, and those with two copies have a 12-fold increased risk.
Interestingly, mice bred with the APOE4 allele are more permissive to HSV-1 spread in the brain, likely because APOE4 also makes the blood-brain barrier less stringent. Following this, a meta-analysis found that APOE4 carriers were about 3-fold more likely to harbor HSV-1 infection than non-carriers. And the presence of the APOE4 allele has been found to potentiate the risk of AD posed by HSV-1 to up to a 17-fold increase.
Third, and perhaps most importantly, the infectious model of AD complements the classic amyloid model. Apparently, Aβ actually exhibits potent antimicrobial activities, including against HSV-1.
Aβ plaques in the brain can trap microbes, thus hindering the further spread of the infection. A landmark 2018 study showed that infecting mice with HSV-1 resulted in Aβ aggregation in the brain. But the generated amyloids bound to and neutralized HSV-1, which prevented herpes simplex encephalitis and prolonged the mice’s lifespan — at the cost of increased brain Aβ, however (Figure 10). Other studies in 2016 and 2024 have also found that Aβ plaques defend against neuronal infections.
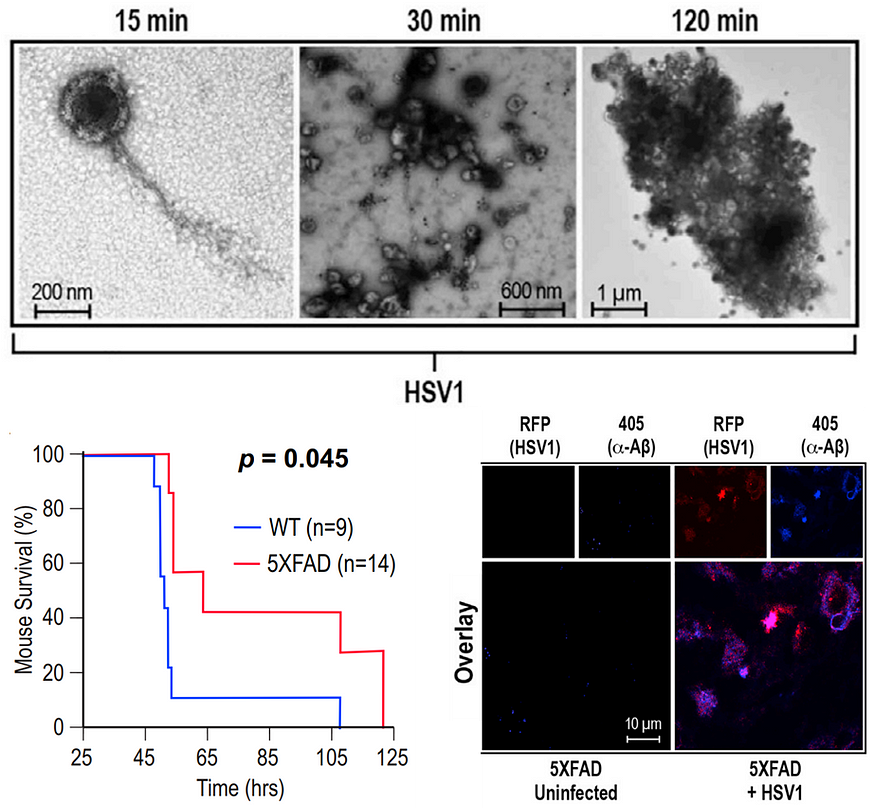
These findings are initiating a paradigm shift in how we view Aβ. We thought they were purely neurotoxic, causing AD. But it seems that Aβ may be a defense mechanism against brain infections. Its primary role could be protecting rather than harming us — a 180° flip in reasoning.
Aβ first evolved about 400 million years ago in vertebrates, and since then, its gene sequence has remained mostly the same. The evolutionary-conserved Aβ may, thus, serve a crucial function over eons, probably pathogen defense. If Aβ only confers a toxic trait, it should be gradually eliminated throughout the course of evolution. But this isn’t the case. There must be a reason why evolution preserves Aβ.
About tau proteins, while some studies have found they exhibit antibacterial activities, not much research has been done on whether they have antiviral properties. That said, some scientists have speculated that tau phosphorylation acts as a danger signal to a foreign invasion.
As a result, some scientists have proposed the antimicrobial hypothesis of AD. In a 2018 review paper by Robert Mori (1961–2019), a former Harvard neurologist who also directed the 2018 landmark study (Figure 10), this hypothesis was articulated for the first time:
In this model, β-amyloid deposition is an early innate immune response to genuine, or mistakenly perceived, immunochallenge. Aβ first entraps and neutralizes invading pathogens in β-amyloid. Aβ fibrillization drives neuroinflammatory pathways that help fight the infection and clear β-amyloid/pathogen deposits. In AD, chronic activation of this pathway leads to sustained inflammation and neurodegeneration. — Moir et al. (2018).
Dr. Moir also emphasized that the antimicrobial hypothesis complements rather than contradicts the classic amyloid model of AD. It still relies on the fact that Aβ plaques and NFTs drive neurodegeneration but adds that their trigger could be the brain’s defense mechanisms against infections.
3.5. Cons of the infectious model of AD
All that said, however, the direction of causation remains unclear.
Some scientists suggest that because AD is mainly a disease of old age, aging itself may make one susceptible to HSV-1 and other infections. After all, aging comes with declining immunity and a more permeable blood-brain barrier, both of which encourage brain infections.
Moreover, HSV-1 has a high global prevalence of 66.6% in those under 50. Yet not everyone infected will develop dementia or AD. But this doesn’t debunk the hypothesis that multiple reactivations of HSV-1 throughout life — under conditions of immunosuppression or stress— contribute to AD. This means a subset of individuals prone to HSV-1 reactivations are the ones susceptible to AD. Indeed, HSV-1 prevalence in the brain is generally higher in individuals with than without AD (Figure 11).
However, this also leads to another inconsistency in the infectious model of AD — not every AD patient has an HSV-1 infection in their brain. The prevalence of HSV-1 positivity in the brain of AD patients hovers at 50–80%, depending on the study (Figure 11). This observation means that HSV-1 may not be the prerequisite trigger of AD (as in the case of EBV/human herpesvirus 4 in multiple sclerosis — more on this later), but it doesn’t deny that it could be a relatively common trigger of AD.

There’s also a lack of clear evidence linking HSV-1 viral load and spread with progressive brain damage in AD. Although one study found that 72% of HSV-1 viral load in the brain was located within Aβ plaques in AD patients at autopsy, no research has definitively demonstrated that HSV-1 triggers plaques and tangles along its infection pathways in human brains, particularly in the early to middle stages of AD.
One reason may be that, if the antimicrobial protection hypothesis is correct, HSV-1 could already be neutralized by Aβ (and perhaps tau), making it difficult to track the virus within the brain.
Studying the virus’s distribution or infection trajectory in the brain is only feasible at autopsy, as opening a living human brain carries obvious consequences. That’s why most such investigations rely on animals, where euthanizing them early allows us to track viral activity in the brain.
…a herpesvirus protein [may] be important for AD development but would not necessarily result in the detection of the complete virus thus providing a potential explanation for the lack of easily identifiable pathogen in the brains of AD patients. Moreover, if Aβ destroys its viral targets as an early or ‘triggering’ event in AD, it may become increasingly difficult to identify viral epitopes late in the disease process, particularly at end of life at autopsy. This could explain to some degree the inconsistencies in findings of virus in AD brains.
— Allnutt and Jacobson (2020).
3.6. Our most likely suspect
Then again, it’s hard to deny all the abovementioned evidence that HSV-1 is a prime contributor to AD. So, the most plausible scenario is that old age and microbes, as well as other factors like genetics, lifestyle, and environment, all work sophisticatedly in instigating AD.
This multifaceted approach is consistent with the adaptive response hypothesis proposed in 2014, where Aβ serves as a defense mechanism to maintain brain homeostasis against various acute (short-term) and chronic (long-term) stressors — such as oxidative stress, inflammation, metabolic stress (e.g., insulin resistance, metal ion dysfunction, lipid dysregulation, etc.), traumatic brain injury, and infections (Figure 12).

In this manner, there might not be a single cause or trigger of AD. But this also means we will never solve the origin trigger of AD. In a way, it may also mean taking the easy way out. There could be a common trigger in some, many, or even most cases of AD. As long as this possibility exists, we shouldn’t give up on pinpointing the origin trigger of AD.
If we can identify a common trigger of AD, efforts to target this trigger would bring more fruitful results than targeting the pathological signs after the initial trigger has been pulled — i.e., plaques and tangles.
An interesting corollary is that a 2022 study has conclusively determined that Epstein-Barr virus (EBV) —i.e., human herpesvirus 4 — is the prerequisite trigger of multiple sclerosis in a series of analyses. Multiple sclerosis is another type of neurodegenerative disease. How this study proved this is nothing short of fascinating, and I’ve written about it here: How a Single Study Proved the Cause of Multiple Sclerosis Is a Virus.
While HSV-1 may not be a prerequisite trigger of AD — given that not 100% of AD patients have been exposed to HSV-1 — it could still be a relatively more common trigger of AD. To better comprehend this, let’s look at the common factors of AD (Figure 13), which include:
Old age (nearly 100% of patients, excluding familial AD)
Female dominance (65% of patients)
APOE4 genetic risk factor (40–65% of patients)
History of traumatic brain injury (10–15% of patients)
Presence of HSV-1 in the brain (50–80% of patients)
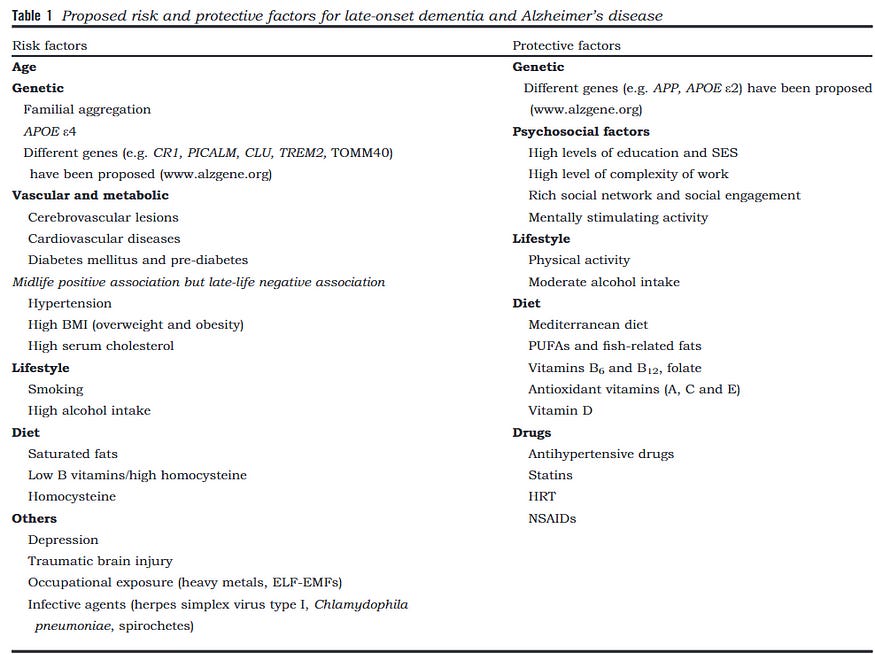
Based on that, our most likely suspect seems to be older females with APOE4 gene and/or HSV-1 brain infection. Of course, the more appropriate way to compare these suspects or risk factors is through risk estimation (i.e., risk ratio, odds ratio, etc.), but then it’s difficult to draw inferences regarding prerequisite or communal trigger in that case.
(It’s unclear why AD affects females more often, with the reasons proposed ranging from hormonal, genetic, and biological differences.)
To wrap it up, the classic amyloid and tau hypotheses of AD are incomplete, with the biggest missing piece being the origin trigger. While AD is undoubtedly a multifactorial disease, there should and likely be a relatively more common trigger that can explain the occurrence of plaques and tangles in the hippocampus, as well as the common pathological signs of disease like oxidative stress and inflammation.
Whoever can pinpoint this elusive trigger — be it HSV-1†, another infectious agent, or some other unknown entity — will be on the verge of solving this seemingly unsolvable disease. Imagine if we could inhibit or vaccinate against this trigger, how much prevalence of AD could we cut?
†As discussed, we don’t have an effective vaccine against HSV-1 as it’s one of the very tricky viruses to vaccinate against. Moreover, since HSV-1 typically causes mild oral sores (and severe encephalitis in rare cases), there hasn't been an urgent need to develop a vaccine for HSV-1.
If you have made it this far, thank you. If you enjoyed this, please subscribe below and share it with others. You can also tip me here—any support is much appreciated!
I must thank you for such a well written and thought out paper delving into one of the most complex disease to occur in aging humans that still evades successful treatment outcomes.