



Solving the Cause of Parkinson’s Disease, As An Academic in This Field
Building on and completing the “gut-first hypothesis” of Parkinson’s disease (PD).

This article was originally published at Microbial Instincts on Feb 15, 2024.
For my Master’s degree in Science (by research), my thesis was on Parkinson’s disease (PD), where I studied its disease mechanism and potential treatments in lab-grown neurons. Naturally, I knew a fair bit about PD in general from my 2–3 years of master’s research.
For those unfamiliar, PD is a motor disorder due to degenerated neurons in the brain that control movement. As a result, PD patients show motor deficit signs such as tremors (shaking) and bradykinesia (slow movement). PD is quite common, being the second most common neurodegenerative disease after Alzheimer’s disease (AD). Muhammad Ali, known as the greatest boxer of all time, for example, suffered from PD.
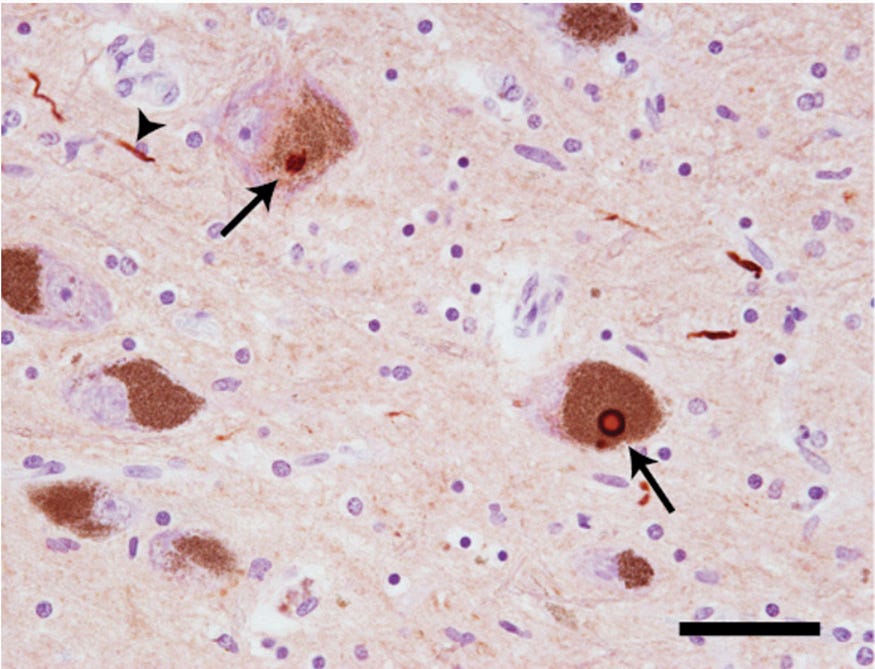
But to this day, the cause of PD remains unclear, and there’s no cure. One of the hot topics about the cause of PD is the gut-first hypothesis, where α-synuclein protein becomes misfolded in the gut and spreads to the brain via the vagus nerve (Figure 2). Misfolded α-synuclein then aggregates into neurotoxic Lewy bodies and neurites (Figure 1). To this day, this topic remains heavily discussed in academia and major news outlets.
As an academic who has studied this disease for years, let me tell you the fascinating story about the gut-first hypothesis and how it fits another lesser-known hypothesis to decode the cause of PD.

The gut-first hypothesis of PD
Professor Heiko Braak, MD, is an ingenious German neuroanatomist known for characterizing the stages of disease progression of Alzheimer’s and Parkinson’s diseases (AD and PD), the two most common neurodegenerative diseases worldwide.
In a 2003 study, Braak et al. performed autopsy analyses on the brain samples of 110 people with PD and 58 healthy people. They found the earliest signs of α-synuclein aggregates were always at (i) the dorsal motor nucleus of the brainstem, where the vagus nerve connects, and (ii) at the olfactory bulb, though less frequent than the brainstem (Figure 3).
The vagus nerve is the longest nerve in the body. It connects the brainstem, the base of the brain, to various organs, including the enteric nervous system of the gastrointestinal tract. This allows for two-way communication between the brain and other organs.

Subsequent studies managed to identify the presence of α-synuclein aggregates in the gut, stomach wall, and lower esophagus, which are all connected to the enteric nervous system.
The enteric nervous system is basically nerves that innervate the walls of the entire gastrointestinal tract. It’s popularly known as the ‘second brain’ because it’s the most complex nervous system outside the brain.
Importantly, signs of α-synuclein aggregates in the gastrointestinal tract began to appear in the early stages of PD, before such aggregates even appeared in the brain. This rules out a common argument against the gut-first hypothesis of PD — that these aggregates still originate in the brain but spread to the enteric nervous system later. Put simply, α-synuclein seems to aggregate in the gastrointestinal tract first, not the brain.
Animal research further supports the gut-first hypothesis of PD. In a 2019 study, scientists from Johns Hopkins University injected misfolded α-synuclein into the stomach and gut of healthy mice. After a month, α-synuclein aggregates were found in the brainstem. By 10 months, such aggregates spread throughout the brain, and the mice exhibited symptoms of motor deficit similar to PD. Cutting the vagus nerve, however, prevented the spread of α-synuclein into the brain (Figure 4).
This study is valuable because it demonstrates cause-and-effect that’s not feasible in humans. After all, it’s unethical to inject a toxic substance into humans. So, this animal study convincingly showed that α-synuclein could spread from the gut to the brain via the vagus nerve.

Even in humans, studies found those whose vagus nerve is cut via vagotomy had a lower risk of developing PD than those with intact vagus nerve. But the effect size of this association is weak, and it doesn’t show whether the protective effect of vagotomy stems from preventing the spread of α-synuclein from the gut to the brain. Nevertheless, it implies the vagus nerve is important in PD development.
(Vagotomy, i.e., cutting the vagus nerve, is a last-resort medical intervention for severe cases of peptic ulcer, which reduces the secretion of stomach acid. But, depending on where it’s cut, vagotomy can cause some side effects, such as slowing peristalsis and decreasing bile and enzyme secretions from the liver, gallbladder, and pancreas.)
If α-synuclein aggregation begins in the gut, gastrointestinal symptoms should predate the onset of PD, which occurs when neurodegeneration in the brain is so extensive that the brain fails to control body movement. Indeed, multiple cohort studies, including a 2023 study, reported that gastrointestinal symptoms such as constipation, swallowing difficulty, and bloating increase the risk of developing PD later.
Overall, the evidence is convincing that the origin of PD begins in the gut. When Braak first proposed the gut-first hypothesis of PD in 2003, not many found it believable. But more than 20 years later, his hypothesis stands strong amidst further scientific scrutiny.
What triggers the misfolding?
Now, the big question is what triggers the misfolding and aggregation of α-synuclein in the gut? Akin to how β-amyloid aggregation is the distinct hallmark of AD, α-synuclein is the same for PD — this much is clear.
Braak hypothesized some sort of an enigmatic virus triggering the first misfolded α-synuclein, which then catalyzes further misfolding and eventually aggregation of α-synuclein that’s toxic to neurons. Alternatively, the virus could spread from the gut to the brain via the vagus nerve, seeding α-synuclein aggregation along the way (Figure 5).
In a 2003 paper, Braak et al. wrote PD “might originate outside of the central nervous system, caused by a yet unidentified pathogen that is capable of passing the mucosal barrier of the gastrointestinal tract.” This unidentified pathogen may then infiltrate the brain via the vagus nerve.
In a subsequent paper in 2007, Braak and colleagues proposed the “dual-hit hypothesis” to account for the early appearance of α-synuclein aggregates in the olfactory bulb in addition to the gut. The olfactory bulb is a brain region where the olfactory nerves connect to the nose.
“We propose that a neurotropic pathogen, probably viral, enters the brain via two routes: (i) nasal, with anterograde progression into the temporal lobe; and (ii) gastric, secondary to swallowing of nasal secretions in saliva,” Braak et al. wrote. “It is concluded that the most parsimonious explanation for the initial events of sporadic Parkinson’s disease is pathogenic access to the brain through the stomach and nose — hence the term ‘dual‐hit’.”

What pathogen or virus is capable of inducing α-synuclein aggregation then? Braak et al. aren’t sure of any specific pathogen but did point out that the influenza A virus (influenza from hereon) could be a culprit.
Influenza has been suspected to be a risk factor for PD for decades. This suspicion stems from the observation that PD incidence soared to 2.5–3% in the US in 1940–1950s after the 1918 influenza pandemic, which returned to baseline at 1–2% in the following decades.
In animal studies, nasal infection with influenza virus can induce α-synuclein aggregation and neurodegeneration in the brain reminiscent of PD. One study mapped the influenza virus’s infection trajectory, detecting the virus in the lungs first, followed by the enteric nervous system and the brainstem and other brain regions typically affected in PD. The virus was also detected in the vagal nucleus of the brainstem, suggesting the virus could use the vagus nerve as a conduit to infect the brain.
Another similar study showed that influenza can infiltrate the olfactory bulb and the brain of mice via the olfactory route as well, triggering α-synuclein aggregation along the way. However, this detrimental outcome can be prevented by administering antivirals beforehand.
(Despite being a respiratory virus, some influenza virus strains can infect the nervous system and gastrointestinal tract, similar to SARS-CoV-2).
Interestingly, animal studies also show that neurons in the key brain regions involved in PD continue to degenerate even after the influenza virus is cleared by the immune system. This may explain the absence of the influenza virus during the brain autopsy of deceased PD patients, as sufficient time had elapsed for the body to clear the virus.
A nationwide study further strengthens this case. Using data from Danish National Patient Registry between 1977 to 2016, a 2021 study identified every single case of PD, which was matched to five controls based on age and sex. They ended up with 10,271 PD patients and 51,355 controls for analyses, revealing that prior influenza diagnosis increased the risk of PD by 1.7-fold in later years, an effect not seen with other pathogens.
A more recent nationwide study in Finland and the U.K. also replicated the association between influenza and increased risk of PD (and AD). I covered this study in detail here: “The Case for Virus Origin of Neurodegenerative Diseases Is Getting Stronger and More Important.”
The influenza virus isn’t the sole suspect of PD. Several cohort studies have found consistent evidence of hepatitis C virus and Helicobacter pylori bacterium increasing the risk of PD in later years. One study found that Covid-19 survivors were also at a 1.5-fold elevated risk of PD. The gut microbiome has also been implicated in PD, although we don’t know what’s the precise microbe in the microbiome that’s responsible for this.
Evidence also suggests the risk of PD from pathogens adds up. A 2015 study found the more pathogens someone is positive for, the higher the risk of PD. For instance, testing positive for five or six pathogens increased the risk of PD by 7.8-fold compared to 1.9-fold when testing positive for one or two pathogens only. The pathogens examined were herpes simplex virus 1, cytomegalovirus, Epstein-Barr virus, B. burgdorferi, C. pneumoniae, and H. pylori. Unfortunately, the influenza virus wasn’t examined.
Compounding all this is the fact that α-synuclein also exhibits anti-microbial properties against clinically relevant pathogens. Deleting the α-synuclein gene in mice did not affect the usual nervous system function, but it made them more susceptible to life-threatening infections. Another mice study showed that α-synuclein colocalizes with viral capsids in neurons to prevent further replication and spread of the virus.
The anti-microbial activities of α-synuclein are an important piece of the puzzle as they suggest that α-synuclein aggregation could be a defense mechanism against pathogens that infect the nervous system.
What accelerates the misfolding?
However, not all individuals infected with the influenza virus or other pathogens will develop PD. There must be another contributing factor that hits the threshold for PD to develop.
During my master’s research, I cultured neurons in the lab. I then exposed them to neurotoxic agents to induce neurodegeneration and see what treatments can mitigate the neurodegeneration. Guess what neurotoxic agent I used? It’s rotenone, a natural compound used in pesticides.
In fact, it’s a common practice in laboratories to use neurotoxic agents to cause PD in animals and cells. Some neurotoxic agents are capable of targeting neurons important in PD, particularly dopaminergic neurons at the substantial nigra that’s responsible for movement control.
In an ingenious yet underappreciated 2017 study, Richard Smeyne, PhD, Chair of the Department of Neuroscience at Thomas Jefferson University, and colleagues showed that prior influenza virus infection could synergize with MPTP neurotoxicant to cause PD in mice. Like rotenone, MPTP is a known PD-inducing neurotoxic agent. Thankfully, influenza vaccination or antivirals were sufficient to thwart the development of PD in the mice.
According to the authors, the finding that MPTP produced greater neurodegeneration in mice previously infected with influenza suggests that “prior influenza infection, even if resolved, could increase the sensitivity of DA neurons to a second insult.” (DA means dopaminergic neurons, the main neurons in the substantia nigra that’re degenerating in PD.)
Smeyne and colleagues then proposed that “influenza infection can act as the first “hit” in a “multi-hit” model [of PD].” This means that influenza may trigger the very first misfolding and aggregation of α-synuclein — the first hit. Subsequently, exposure to environmental toxins like pesticides could be the next hit in triggering further aggregation of α-synuclein.
Chronic exposure to pesticides containing neurotoxic agents such as rotenone and paraquat explains why farmers are about 2.5-fold more likely to develop PD than non-farmers. Such pesticides are still being sold, and thousands of farmers with PD have sued pesticide manufacturers in the U.S. for prioritizing sales over communication of safety.
Other hits in PD development may include stress, physical inactivity, and improper diet. These factors are generally detrimental to health and contribute to all sorts of diseases by imposing a state of chronic inflammation and oxidative stress on the body, which are also favorable conditions for α-synuclein to aggregate (Figure 6).

Genetics are also important in PD, although they can’t fully explain PD. Only about 15% of PD patients possess genetic risk factors for PD, such as the mutated PARK2 gene, which makes the protein parkin that helps degrade and recycle proteins. Other examples are PINK1 and LRRK2 genes, which code for protein kinases that protect the mitochondria.
One interesting study in 2019 showed that intestinal infection in mice lacking the PINK1 gene resulted in the development of PD compared to normal mice with the PINK1 gene — suggesting that genetic factors may also influence the impact of pathogens on the risk of PD.
It’s also possible that environmental, lifestyle, or genetic factors may serve as the first hit of PD rather than pathogenic agents. In this case, pathogens can still be the next hit in the multiple-hit hypothesis of PD.
Solving the cause of PD
The world has always been full of microbes, and they greatly influence the evolutionary trajectory of other living organisms, including humans. The gut microbiome is a prime example of this, where trillions of microbes reside therein in an intricate balance with the immune system.
The two most vulnerable body sites to the outside environment are the olfactory (inhale) and gastrointestinal (eat) tracts (Figure 6). Pathogens that manage to infiltrate the nervous system via these routes may trigger the misfolding and aggregation of α-synuclein as a defense mechanism. This may serve as the first hit in the threshold of PD development.
This is also consistent with the fact that PD is an age-related disease, which typically affects those over 60 years old. As we get older, our immune system naturally weakens — due to wear and tear like any other organ — making us prone to the onslaught of pathogens.
However, not all influenza patients will become PD patients. Subsequent hits — such as environmental, lifestyle, and genetic factors — are still needed to push the PD threshold even further and cause the disease.
The findings that influenza vaccination or antivirals can prevent PD in the animal studies I’ve discussed above are encouraging. But there’s a lack of data on how influenza vaccination affects the risk of PD in humans for some reason, probably due to the low awareness in this area. In AD, however, a meta-analysis has reported that those who received at least four annual influenza vaccines had a 49% lower risk of dementia.
Although it’s anti-climatic, there isn’t a single cause of PD, at least based on what’s currently known. In the end, the “multiple-hit” hypothesis appears to be the most plausible explanation of the cause of PD.
What do you think?
If you have made it this far, thank you. If you enjoyed this, please subscribe below and share it with others. You can also tip me here. :))
The multi-hit hypothesis does seem reasonable, in that many other diseases (eg CoVid-19) affect people with co-morbidities more than otherwise healthy people. Of course actually showing an underlying mechanism for this, as you've tried to do here is really difficult.
Thanks for writing this up.